18F标记放射性药物的方法与新技术
放射性药物是实现疾病早期诊断和治疗的基础,在神经系统、心血管、肿瘤、乏氧组织、基因显像等方面具有广阔的应用前景。正电子发射断层显像(PET) 具有高灵敏度和高分辨率。这一技术需要合适的正电子核素标记的药物。PET 显像可以采用18F、11C、15O、13N、68Ga、64Cu等核素,其中18F是使用最广泛的核素。但由于18F的半衰期比较短,必然要求18F显像剂能快速标记和分离。18F-FDG(18F-氟代脱氧葡萄糖) 是研究最多的肿瘤显像剂,已经广泛应用于临床PET 显像。但迄今为止,18F 标记的PET 显像剂种类仍然很少,18F标记往往步骤较多,导致合成的标记物放射性活度低。因此,新的PET 显像剂的研发受到人们的广泛关注。本文介绍了近几年18F标记放射性药物的新方法与新技术,包括18F脱标识法、18F-FDG 直接标记多肽、Al18F络合标记、水溶液中18F-氟硼酸酯的标记、基于微流控芯片的PET 药物标记合成技术等,旨在为有关研究者提供参考。
1 18F脱标识法
脱标识法(detagging) 是用目标分子结合标识物,依据标识物的性质进行纯化或检测,然后再将目标分子和标识物分离。目前,新的18F脱标识法与传统的脱标识法都是基于氟固相萃取而进行分离纯化的。氟固相萃取( fluorous solid phase extraction,FSPE)是一类特殊的固相萃取技术,用氟化硅作为载体,将含全氟链的氟化物和其他有机化合物的混合物上载到柱子上,先用憎氟溶剂洗脱,得到其它有机化合物,再用亲氟溶剂洗脱,得到含全氟链的氟化物。氟固相萃取技术是一种高效,快速的分离方法。
传统的放射性标记脱标识法中,放射性标记和脱标识是分两步进行的。新的18F脱标识法与传统的脱标识法不同,氟化本身就是一个脱标识过程,这样只有发生了氟化反应的化合物才能够洗脱下来,得到的都是高比活度标记化合物(图1)[1]。Gouverneur 首先用F亲电取代的方法进行脱标识[2],如图式1所示,带有全氟链的硅基团上带有烯丙基,在Grubbs 催化剂的催化下,可以和末端烯烃发生烯烃复分解反应,在亲电氟化试剂Selectfluor的作用下,在氟化反应的同时脱去硅基团。利用氟固相萃取就可以将生成的氟标记物和含全氟链的原料进行分离。


Gouverneur发展了氟亲核取代的脱标识法[1],含有全氟链的磺酸环氧乙烷化合物,在18F负离子的进攻下,发生亲核取代反应,制备18F环氧乙烷合成子,放化产率大于89% ,用氟固相萃取柱快速纯化,再和二硝基咪唑反应,可以制备PET显像中常用的[18F]FMISO,放化产率53%(图式2) 。

2 18F标记多肽
大量研究表明,多肽与受体的结合具有高特异性、高亲和性、细胞膜穿透性好、清除速度快等优点。但迄今为止,18F标记多肽的方法不多。通常需要合成前体化合物,再经过标记、偶联、分离纯化等多个步骤。这种方法反应时间长、产率低、化学选择性差。
2. 1 18F-FDG 直接标记多肽
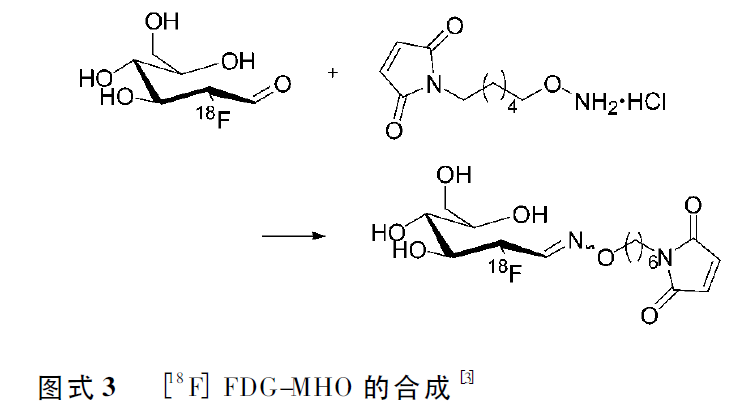
2008 年,Wuest等[3]报道了利用18F-FDG的醛基和氨氧基缩合反应形成肟基,合成出一种新的合成子[18F]FDG-MHO([18F]FDG-maleimidehexyloxime),用于多肽和蛋白质的18F标记(图式3)。[18F]FDG 和N-(6-氨氧基-己基)-马来酰亚胺溶解在80%乙醇中,加热到100℃,密闭反应15min。在100℃时,FDG的构象在环状和线型之间达到动态平衡,而线型态的FDG在1位显示出醛基的功能,在一定条件下,可以与氨氧基反应。反应物经HPLC 纯化后,[18F]FDG-MHO 放射性产率为45%—69%,整个合成与纯化时间缩短到45min内。[18F]FDG-MHO 标记三肽GSH和蛋白质anxA5的实验表明,[18F]FDG-MHO 的化学选择性和标记效率有一定提高。温和条件下,标记反应10min,GSH浓度在1mg—1μg/ml,[18F]FDG-MHO-GSH 的产率大于95%,即使低浓度的GSH(100ng/ml,10ng/ml) ,其产率(92%,47%)比以往报道的[18F]FBAM产率(20%,5%)高很多。与多肽相比,蛋白质anxA5 的标记时间较长(60min),由于蛋白质中只含有一个半胱氨酸标记位点,标记的选择性专一,标记率为43%—58%。低蛋白浓度(100μg)下,与[18F]SFB的标记率(10%)相比,[18F] FDG-MHO对anxA5的标记率相对较高。
2009 年,Mohammad 等直接利用开环[18F]FDG上的醛基与氨氧基化的多肽分子偶联,一步反应完成18F标记[4](图式4)。[18F]FDG-RGD和[18F]FDG-cyclo( RGDDYK) 的放射性标记产率分别为28%和41%,HPLC分析显示副产物少。当温度控制在100℃,pH=1.5—2.5,可以得到较高的产率;pH值高于4,产率极低。但这种高温低酸度的条件对于多肽小分子较适合,对于长的多肽或者蛋白质就不太适用了。同时,标记物中存在肟的E、Z两种构象,难以分离。生物检测表明,[18F]FDG-cycloRGDDYK )的IC50值为0.67 ± 0.19μM,cyclo( RGDDYK)的IC50值为0.23μM,说明18 F标记后对cyclo( RGDDYK)的生物活性影响不大。同时PET成像也表明,[18F]FDG-RGD 和[18F]FDG-cyclo( RGDDYK)在表达αvβ3的肿瘤中有一定摄取。

Wuest等[5]利用具有化学选择性的肟化反应,用[18F]FDG 对氨氧基化的不同多肽进行标记,标记产率与多肽分子形式有关。高浓度的多肽单体和二聚体标记率较高; 低浓度时,单体更具有明显优势。多肽四聚体则无论浓度高低,标记产率都较低。说明这种直接标记的方法比较适合于小的多肽分子,对于复杂的多肽分子或者蛋白质标记并不适用。要满足较高的标记率,不仅要提高底物用量,对分离纯化也有进一步的要求。
点击化学(click-chemistry) 由于反应快捷,反应位点准确,引起了广泛关注。Maschauer等[6]合成了一系列[18F]FDG 前体化合物,利用点击化学对多肽分子进行标记。其温和的18F标记条件,可以使试剂的稳定性增加,反应后HPLC 分离更容易。但受空间位阻和静电效应,只有部分化合物标记率较高。最近,他们再次利用点击反应,完成对神经降压素系列类似物的18F标记[7]。标记率和放化纯度都较高,生物稳定性及活性都有新的改善(图式5)。与临床使用的[18F]galacto-RGD (标记率10%,时间200min) 相比,该方法使标记率提高一倍,同时大大缩短了合成时间(70min)。其中[18F]FGlc-NT4肿瘤摄取较好,有望用于活体PET 显像。

2. 2 Al18F络合标记多肽
利用点击化学(click-chemistry) 将炔和叠氮化合物进行环加成反应,也可以进行多肽的18F标记[8,9]。虽然点击化学标记率高(大于80%),但是该方法需要多步合成和分离,耗时长。另外,在生物分子上引入硅基团也可以实现18F一步标记。但是所形成的标记物不稳定,必须在硅上引入特丁基,修饰基团增大了化合物的脂溶性,同时降低了化合物和靶标的特异性结合[10]。
2009 年,Laverman 等发展了一种新的18F标记多肽的方法[11,12]。18F-KF 和AlCl3反应形成Al18F,然后和连接在多肽上的NOTA 配体发生络合反应(图式6) ,形成标记络合物。这是一个两步、一锅(2-step,1-pot) 的反应,在45min内,完成多肽化合物的18F标记和HPLC分离,标记产率可以达到50%,是一种高效、快速的18F标记多肽的方法。Laverman研究了多肽的DTPA 和NOTA 修饰化合物和Al18F的配位,发现多肽DTPA的Al18F络合物在体外血清实验中不稳定,这主要是由于Al18F和DTPA 形成的络合物不稳定,因此选用NOTA 作为络合基团。Ga、In、Zr、Lu和Y的18F化合物也可以和多肽NOTA 修饰化合物络合,但是产物不如Al18F稳定。Al 18F络合多肽NOTA 修饰化合物在体外血清实验中,具有很好的稳定性。用Al 18F络合多肽NOTA 修饰化合物方法对奥曲肽进行标记,18F-NOTA-奥曲肽的亲和性和111In-DTPA-奥曲肽相似。表明该方法对多肽的亲和性影响很小

3 水溶液中18F-氟硼酸酯的标记
对于体积大、多功能团、水溶性或者是温度敏感型的生物大分子,一般很难进行18F标记。亲电氟化试剂( Xe[18F]2、[18F]2、N-氟化类) 由于活性太高,化学选择性差并且通常要求无水的反应条件,限制了大部分水溶性生物分子的标记。此外,亲电氟化很难得到高比活度标记物。虽然采用亲核氟化试剂可以得到高比活度的标记物,但是在水溶液中它们以惰性的水合阴离子形式存在,亲核性差,反应难以进行。
硼酸及其酯类化合物广泛应用在有机合成和生物有机化学研究中,也可以用于PET 显像剂标记前体的合成。和以往C—F 键的形成方法不同,硼原子由于亲氟性能够直接将水溶液中氟捕获成键。研究已证实,当水溶液中存在氟化物时,芳基硼酸酯转变成相应的三氟硼酸盐(式1)。Ting等发现硼酸酯类化合物可以作为[18F]-PET试剂的标记前体,是一种新的18F标记方法[13—15]。

4 基于微流控芯片的18F标记技术
微流控芯片,或微流体反应器,是一类由构建在固体基质上的微米级孔道网络构成的微型集成设备,可实现对微流体的精确控制,从而在微尺度下以连续流动的方式进行化学或生化反应。微流控技术充分利用了微流体的特点( 例如远低于宏观流体的雷诺数),结合微型化反应器在传质和传热上的优势,在反应速率、试剂消耗、产率、选择性等方面优于宏观反应器。此外,微流控反应系统还具有重复性好、易于模块化和自动化等突出优势。目前,微流控芯片技术已经广泛应用于多种化学反应和生化分析。
由于PET药物常用的放射性核素如18F和11C等的半衰期较短,通常要求标记反应快速完成,微流控芯片技术很多方面可以满足要求,例如更短的反应时间,更高的放射化学产率和放射化学纯度以及更高的比活度等。此外,除了在标记反应方面的优势,作为微型化的集成反应系统,微流控芯片还具有易屏蔽(无需全尺寸热室)、前体要求低( 可用较低的活度及更少的用量)、易分离纯化等优势。
4. 1 微流控芯片用于[18F]FDG合成
[18F]FDG是目前临床中应用最广泛的PET药物,因而其合成效率的提高备受关注。自从2005年Lee等[16]首次报道采用微流控芯片实现[18F]FDG的标记合成以来,已有多种不同的微流控芯片被用于此种药物的合成研究。采用微流控芯片技术可在15min内完成反应,而商用合成仪大约需50min,大大缩短了标记时间,可使用较低活度的前体,产物的放射化学产率和放射化学纯度都有不同程度的提高。特别是Lee等采用的方法,从反应体积和自动化程度的角度来看,属于“真正的”微流控芯片,并可实现单人剂量的定制合成。最近,Elizarov等[17]报道的圆形反应器芯片,解决了第一代微流控芯片的反应容量问题,并有望用于制备用现有常规方法合成产率较低的PET 药物。
4. 2 采用微流控技术的其他PET药物合成
除了[18F]FDG 之外,也有很多其它PET 药物采用微流控芯片技术合成的报道。Lu等[18]采用水力学驱动的微型反应器合成了11C和18F标记的几种羧酸酯。Briard等[19]通过一步氟化,合成了N-[18F]氟代乙酰基-N-(2,5-二甲氧基苄基) -2-苯氧基苯胺,一种高亲和力的转运蛋白配体。Chun等[20]报道了邻位取代[18F]氟代芳烃的快速合成。Saiki等[21]报道了采用一次性的微流控芯片进行无载体的[18F]氟化物的电化学浓缩。
5 总结和展望
PET 显像剂已经成为放射性药物化学的研究热点,发展18F标记的新方法和新技术具有重大的意义。本文介绍了近几年来的研究进展:18F脱标识法,氟化过程本身就是脱标识过程,这样就只有发生了氟化反应的化合物能够洗脱下来,得到的都是标记化合物。用该方法可以得到高比活度的标记物,并能用固相萃取柱实现快速分离。由于大部分PET中心都有18F-FDG合成仪,这非常有利于18F-FDG 进一步直接标记多肽的应用。Al18F络合标记多肽,利用Al18F和NOTA形成络合物,这是一个“两步一锅”的反应,大大缩短了多肽标记和分离的时间。利用18F-氟硼酸酯,可以实现水溶液中18F-标记。基于微流控芯片的PET药物标记合成技术,使18F-标记向自动化和智能化发展。以上这些标记方法都具有里程碑式的意义,也具有重要的参考价值。
[1] Bejot R, Fowler T, Carroll L, Boldon S, Moore J E, Declerck J, Gouverneur V. Angew. Chem. Int. Ed., 2009,48: 586—589
[2] Boldon S, Moore J E, Gouverneur V. Chem. Commun.,2008, (31): 3622—3624
[3] Wuest F, Berndt M, Bergmann R, Hoff J V D, Pietzsch J. Bioconjugate Chem., 2008, 19: 1202—1210
[4] Namavari M, Cheng Z, Zhang R, De A, Levi J, Hoerner J K, Yaghoubi S S, Syud F A, Gambhir S S. Bioconjugate Chem.,2009, 20: 432—436
[5] Wuest F, Hultsch C, Berndt M, Bergmann R. Bioorg. Med. Chem. Lett., 2009, 19:5426—5428
[6] Maschauer S, Prante O. Carbohyd. Res., 2009, 344: 753—761
[7] Maschauer S, Einsiedel J, Haubner R, Hocke C, Ocker M, Hübner H, Kuwert T, Gmeiner P, Prante O. Angew. Chem. Int. Ed., 2010, 49: 976—979
[8] Li Z B, Wu Z H, Chen K, Chin F T, Chen X Y. Bioconjugate Chem., 2007, 18: 1987—1994
[9] Marik J, Sutcliffe J L. Tetrahedron Lett., 2006, 47: 6681—6684
[10] Mu L J, Hhne A, Schubiger P A, Ametamey S M, Graham K, Cyr J E, Dinkelborg L, Stellfeld T, Srinivasan A, Voigtmann U, Klar U. Angew. Chem. Int. Ed., 2008, 47: 4922—4925
[11] McBride W J, Sharkey R M, Karacay H, D'Souza C A, Rossi E A, Laverman P, Chang C H, Boerman O C, Goldenberg D M. J. Nucl. Med., 2009, 50: 991—998
[12] Laverman P, McBride W J, Sharkey R M, Eek A, Joosten L, Oyen W J G, Goldenberg D M, Boerman O C. J. Nucl. Med., 2010, 51: 454—461
[13] Ting R, Harwig C, Keller U, McCormick S, Austin P, Overall C M, Adam M J, Ruth T J, Perrin D M. J. Am. Chem. Soc., 2008, 130: 12045—12055
[14] Ting R, Adam M J, Ruth T J, Perrin D M. J. Am. Chem. Soc., 2005, 127: 13094-13095
[15] Harwig C W, Ting R, Adam M J, Ruth T J, Perrin D M. Tetrahedron Lett., 2008, 49: 3152-3156
[16]Lee C C, Sui G, Elizarov A, Shu C J, Shin Y S, Dooley A N, Huang J, Daridon A, Wyatt P, Stout D, Kolb H C, Witte O N, Satyamurthy N,Heath J R, Phelps M E, Quake S R, Tseng H R. Science, 2005, 310: 1793—1796
[17] Elizarov A M, van Dam R M, Shin Y S, Kolb H C, Padgett H C, Stout D, Shu J, Huang J, Daridon A, Heath J R. J. Nucl. Med., 2010, 51: 282—287
[18] Lu S Y, Watts P, Chin F T, Hong J, Musachio J L, Briarda E, Pike V W. Lab Chip., 2004, 4: 523—525
[19] Briard E, Zoghbi S S, Siméon F G, Imaizumi M, Gourley J P, Shetty H U, Lu S, Fujita M, Innis R B, Pike V W. J. Med.Chem., 2009, 52: 688—699
[20] Chun J H, Lu S, Lee Y S, Pike V W. J. Org. Chem., 2010, 75: 3332—3338
[21] Saiki H, Iwata R, Nakanishi H, Wong R, Ishikawa Y, Furumoto S, Yamahara R, Sakamoto K, Ozeki E. Appl. Radiat. Isot., 2010, 68: 1703—1708
相关文章: